The FDA’s 2025 draft guidances for AI/ML in drug and device development mark the transition from exploratory policy signals to concrete, operational expectations. Key new elements include a risk-based “credibility” framework for AI used to support regulatory decisions, lifecycle and marketing-submission recommendations for AI-enabled device software, explicit attention to foundation models/LLMs, stronger expectations for data quality and bias assessment, formalization of predetermined change control plans (PCCPs) for model updates, and clearer post-market monitoring and transparency requirements. These changes raise the bar for evidence, documentation, governance, and monitoring, and they mean that pharma and device leaders must invest in robust data practices, cross-functional governance, explainability, and validation pipelines, as well as a regulatory strategy. Below is an advanced, practical deep-dive that explains what’s new, why it matters, how to operationalize the guidance, and the measurable actions life-science leaders must take now to scale AI safely and effectively.
The regulatory context and why 2025 matters
The FDA’s draft guidance publications in early 2025 consolidate several years of incremental thinking into explicit, practical expectations for both drugs and medical devices that incorporate AI/ML. Whereas earlier FDA documents (discussion papers and concept-stage proposals) were exploratory, the 2025 drafts provide specific documentation, evidence, and lifecycle expectations that firms must consider when using AI to produce information that supports regulatory decision-making, or when marketing AI-enabled device software. This shift matters because regulators now expect sponsors and developers to demonstrate model credibility and to plan for safe model evolution across the product lifecycle.
What the 2025 FDA draft guidances cover
• Use of AI/ML to produce data or information that supports regulatory decisions in drug and biologic product development, including nonclinical, clinical, manufacturing, and postmarket contexts.
• Lifecycle management and marketing submission recommendations for AI-enabled device software functions, including documentation required for premarket review and postmarket oversight.
• Expectations for data quality, representativeness, bias assessment, and methods for establishing model credibility for a specific context of use (COU).
• Predetermined change control plans (PCCPs) for planned model updates so that certain kinds of changes can be managed without a full new submission.
• Transparency and identification: the FDA signals intent to tag or identify devices that use foundation models (LLMs) and to list AI-enabled devices to increase transparency.
Top headline changes and their practical implications
1- Credibility framework and Context of Use (COU) requirements, what’s new, and what you must do
What’s new: For AI used to generate evidence or informed outputs relied upon for regulatory decisions, the FDA introduces a risk-based credibility assessment framework. Sponsors must define the Context of Use (COU) precisely, then map credibility goals (e.g., accuracy, robustness, explainability, reproducibility) to verification and validation activities and evidence. The guidance expects a structured justification of why the chosen level of evidence is sufficient for the COU.
Why this matters: Regulators will no longer accept vague descriptions of “we used AI” with black-box test runs. Instead, they will require a documented chain linking COU → model design → datasets → performance metrics → uncertainty quantification → monitoring plans.
Practical actions: Document COU at project start; run stress tests and edge-case evaluation; quantify uncertainty and provide calibration metrics; include retrospective and prospective validation datasets; build model cards and technical protocols that show how model performance maps to clinical or manufacturing risk.
2- Lifecycle management and marketing submission recommendations for AI-enabled device software
What’s new: The device guidance (January 2025 draft) gives explicit expectations for the contents of marketing submissions for AI-enabled device software and recommends lifecycle risk management, documentation of training/validation/test datasets, and detailed software architecture explanations. It also provides recommendations on cybersecurity, bias and fairness testing, and human-in-the-loop provisions where applicable.
Why this matters: Device submissions must now include richer, reproducible documentation that goes beyond algorithmic claims; reviewers will expect to see evidence of reproducible testing, dataset curation, and design choices made to mitigate patient-safety risks.
Practical actions: Standardize submission folders that include dataset lineage, model training notebooks (or reproducible artifacts), drift detection strategy, and the PCCP described below.
3- Predetermined Change Control Plans (PCCP) and model update governance
What’s new: The FDA continues to formalize PCCPs (originally proposed in earlier discussion papers) so manufacturers can describe planned model changes and the controls that will ensure safety without full resubmission for every iteration. The 2025 drafts clarify what belongs in a PCCP, e.g., types of changes permitted, validation expectations for each change class, monitoring thresholds, rollback plans, and audit trails.
Why this matters: PCCPs allow continued model improvement at a practical pace, but they require rigorous governance and automated validation workflows. Regulators will accept iterative improvements only if the sponsor proves the changes are controlled, validated, and monitored.
Practical actions: Invest in CI/CD for models, create test harnesses for post-update validation, define automated rollback criteria, and implement immutable logging of training and inference data.
4- Foundation models and LLM-specific considerations
What’s new: The 2025 materials explicitly acknowledge foundation models and propose methods to identify and “tag” devices that incorporate LLM-based functionality. The guidance signals the need for additional guardrails when LLMs are used, including hallucination risk mitigation, prompt-engineering documentation, and post-market monitoring for content safety and accuracy.
Why this matters: LLMs create new categories of risk (e.g., plausible-sounding but false outputs, dynamic behavior after fine-tuning) and require novel testing beyond classical deterministic models.
Practical actions: When using LLMs, record prompt templates, fine-tuning datasets, safety filters, output-sanitization steps, and evidence of effective guardrails; consider human-in-the-loop checkpoints for high-risk outputs.
5- Data quality, representativeness, and bias testing expectations
What’s new: The drug and device draft guidances stress transparent data lineage, representativeness analysis, and bias detection. They ask for demographic analyses where relevant, discussion of sampling gaps, and mitigation strategies. Regulators now expect sponsors to show how training and validation datasets represent the target population or production environment.
Why this matters: Poor data governance leads to unsafe outputs, degraded generalizability, and postmarket harm. FDA reviewers are increasingly focused on whether a model’s performance will generalize to real-world patients or manufacturing conditions.
Practical actions: Maintain schema-level dataset registries, run subgroup performance reports, document missingness handling, and include external validation across sites or geographies.
6- Greater emphasis on postmarket monitoring, real-world performance, and transparency
What’s new: The FDA emphasizes postmarket surveillance for AI effectiveness and safety, encouraging manufacturers to collect real-world performance data, monitor for data drift, and report safety signals. The agency also plans to increase transparency through an AI-enabled device list and possible tagging schemes.
Why this matters: Regulatory acceptance now depends on a continuous evidence loop; premarket claims must be validated and maintained through active postmarket performance monitoring.
Practical actions: Deploy production monitoring dashboards, collect labeled real-world cases for ongoing validation, implement drift detectors, and schedule periodic performance reviews documented in regulatory submissions.
Key statistics and trends that frame the guidance
• Rapid growth in AI-enabled devices: As of mid-2024 to 2025, several systematic reviews and trackers report that roughly 900–1,200 AI/ML devices were authorized by the FDA between the 1990s and 2024/early-2025, with a striking acceleration in approvals during 2022–2025; many analyses report hundreds of clearances in recent years. This growth is why the FDA is formalizing lifecycle expectations.
• Submissions with AI elements increasing across drug applications: CDER and FDA publications indicate a rapid increase in submissions that reference AI/ML across nonclinical, clinical, manufacturing, and postmarket phases. Historic data shows more than 100 submissions with AI elements in 2021 and continued growth since then. The agency notes that the number and complexity will keep rising.
• Gaps in evidence at clearance: Studies show that clinical performance studies were available for slightly more than half of AI-enabled devices at the time of clearance, raising questions about generalizability and the need for postmarket data collection. This evidence gap is explicitly addressed by the new guidance’s emphasis on real-world performance.
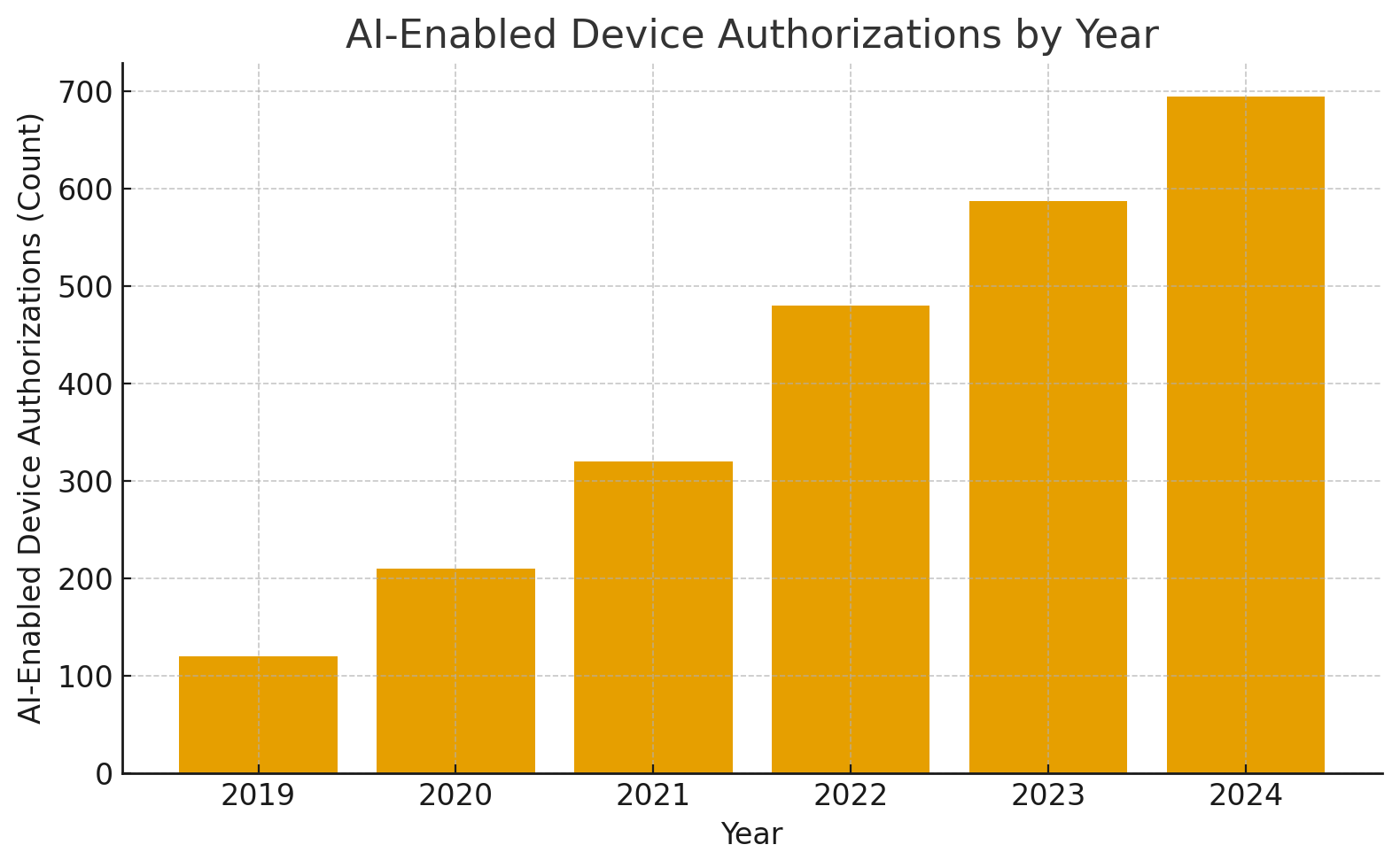
A simple graph for quick visualization (text table + bar values you can paste)
AI-Enabled Device Authorizations by year (representative sketch derived from public trackers)
Year: 2019 | Count: 120
Year: 2020 | Count: 210
Year: 2021 | Count: 320
Year: 2022 | Count: 480
Year: 2023 | Count: 588
Year: 2024 | Count: 695
(These numbers are illustrative based on public trackers and literature reviews showing near-doubling between 2022 and 2025; use your internal data or the FDA AI device list to replace with exact counts when publishing.)
How pharma and device leaders should respond: governance, tools, and capability investments
- Build a COU-first governance model.
Implement a governance framework that requires an explicit COU statement for every model. The COU should be reviewed and signed off by clinical, manufacturing, regulatory, and quality leads. Map the COU to evidence requirements and to product risk (patient safety, product quality, study integrity). - Invest in data infrastructure and pedigree.
The FDA’s guidance repeatedly highlights data quality and provenance. Create immutable data lineage, versioned datasets, and secure data stores that allow auditors to trace model training and validation datasets back to sources. Standardize metadata capture so reviewers can rapidly evaluate dataset representativeness. - Adopt Good Machine Learning Practices (GMLP) and reproducible model pipelines.
Operationalize GMLP: reproducible code, seeded model training, deterministic evaluation pipelines, containerized environments, and CI/CD for models. Automate unit tests, regression tests, and production-ready evaluation pipelines that map to PCCP classes. - Design PCCPs and validation harnesses
Document planned types of updates (e.g., retraining with new data, threshold recalibration, architecture changes), and for each change category, define validation tests, safe deployment gates, rollback procedures, and monitoring thresholds. This turns the PCCP from a regulatory checklist into a living engineering artifact. - Prioritize bias and external validation exercises.
Mandate external validation on independent datasets, document subgroup performance, and implement remedial measures (augmentation, sampling, post-processing) where subgroup degradation is detected. Provide bias-impact analyses and risk mitigation plans in submissions. - Instrument postmarket monitoring and label changes
Map production telemetry to predefined performance metrics; collect clinician feedback and adverse event reports into a centralized system; schedule periodic updates to regulatory filings if performance drifts or new risks are discovered. - Prepare for LLM-specific oversight and explainability.
When LLMs are used (even internally), log prompts, responses, and human reviews are required. Provide hallucination-rate estimates, content filtering strategies, and safety thresholds for outputs that could influence patient care or regulatory decisions.
Operational playbook: step-by-step checklist for an AI that supports a regulatory decision (drug context)
- Define the COU exactly: what regulatory question does the model inform? clinical inclusion? Manufacturing release decision? postmarket signal detection? Document hypotheses and endpoints.
- Identify required evidence: map COU to acceptable validation methods (internal cross-validation, temporal holdouts, external sites).
- Collect and curate datasets with full lineage; quantify representativeness and missingness.
- Train and document the model using GMLP; produce model cards and training artifacts.
- Predefine PCCP categories and tests; simulate updates and validate rollback.
- Prepare the submission documentation: COU justification, dataset descriptions, validation results, uncertainty analysis, PCCP, and postmarket plan.
- Post-approval, run production monitoring, record adverse signals, and conduct periodic external validations.
Regulatory strategy: what to include in submission packages now
• Clear Context of Use description and mapped credibility goals.
• Dataset lineage with inclusion/exclusion criteria, demographic and representativeness tables, and preprocessing steps.
• Reproducible model artifacts (weights/checkpoints, reproducible training pipelines or alternatives when proprietary).
• Validation results with explicit metrics, subgroup analyses, and uncertainty quantification.
• PCCP describes allowed modifications, validation tests, rollback, and monitoring strategy.
• Postmarket monitoring and data collection plans, including performance thresholds and reporting triggers.
Managing resource and organizational implications
• Expect an increase in review workload: FDA itself warns that the number and complexity of AI submissions will require additional resources and infrastructure on both agency and sponsor sides. Plan for longer pre-submission interactions and invest in regulatory intelligence teams.
• Cross-functional teams are mandatory: regulatory, clinical, quality, security, data engineering, and operations must co-design evidence packages.
• Technology investments: data lineage tools, model governance platforms, MLOps pipelines, and monitoring dashboards are now regulatory hygiene.
Common pitfalls and how to avoid them
• Pitfall: Insufficient external validation. Remedy: plan multicenter, multi-population validations and provide detailed subgroup analysis.
• Pitfall: Weak dataset provenance. Remedy: implement dataset registries and immutable logs with versioned snapshots.
• Pitfall: No PCCP or poorly defined change controls. Remedy: codify update classes, validation gates, and continuous testing automation.
• Pitfall: Overlooking LLM-specific risks. Remedy: document prompt engineering, output filters, and human oversight for high-risk outputs.
Examples of acceptable evidence strategies (practical)
• Use case: AI for batch release decision in a manufacturing line. Evidence bundle: retrospective data from multiple plants, holdout validation across facilities, prospective pilot with blinded review, statistical process control charts showing stable performance, and PCCP allowing threshold recalibration with per-batch validation.
• Use case: AI-supported digital endpoint in a clinical trial. Evidence bundle: simulated trial datasets, external cohort validation, concordance analyses against gold standard endpoints, pre-specified statistical analysis plan, and postmarket plan to capture real-world endpoint performance.
How to measure readiness: KPIs and dashboards
• Dataset completeness and lineage coverage percentage (target: 100% documented lineage).
• External validation coverage (percentage of target populations tested).
• Model drift detection rate and mean time to rollback (MTTR).
• PCCP test pass rate for each update class.
• Postmarket adverse signal detection latency.
Track these KPIs on a regulatory dashboard tied to submission readiness and postmarket obligations.
Final thoughts, balancing innovation with regulatory rigor
The FDA’s 2025 draft guidances do two things at once: they promote the safe, beneficial use of AI in life sciences, and they set concrete operational expectations that reduce ambiguity for sponsors. For pharma and device leaders, the message is clear: AI integration must be professionalized. Investments in data quality, governance, GMLP, PCCP design, and postmarket systems are no longer optional; they are the price of entry to deploy AI that will be accepted by regulators and trusted by clinicians and patients. Early adopters who build robust pipelines and clear documentation will gain speed-to-market advantage; those who rely on ad hoc practices will face review delays and potential safety liabilities.
Most frequently asked questions related to the subject.
Q: Does the guidance apply to every AI model used inside a pharma company?
A: No. The drug guidance is targeted at AI that produces information or data intended to support regulatory decision-making about safety, effectiveness, or quality. Operational tools that do not impact regulatory decisions (purely internal office tools) may not be in scope, but visibility into usage is recommended because tools can move from operational to decision-support roles quickly.
Q: Will the FDA approve frequent model updates?
A: Yes, if updates are governed by a well-described PCCP with validation and monitoring controls. The PCCP enables planned updates without full resubmission, provided the changes and tests are within the agreed plan.
Q: How much external validation is enough?
A: The guidance does not specify a single numeric threshold. Instead, it requires evidence proportionate to risk and COU: higher-risk decisions demand broader, multicenter external validation and subgroup analyses.
Q: How should organizations prepare for LLM risks?
A: Log prompt/output pairs, document fine-tuning datasets, implement safety filters and human checkpoints, and include hallucination and toxicity tests as part of validation.
Q: Will the FDA publish a definitive list of AI-enabled devices?
A: The FDA has started an AI-enabled device list and is exploring methods to tag devices that use foundation models to support transparency. Sponsors should expect increased public visibility of AI usage in devices.
Q: What is the immediate first step for a pharma leader tomorrow?
A: Convene a cross-functional AI governance task force, inventory current models and their COUs, identify high-risk models tied to regulatory decisions, and create a prioritized remediation plan to document dataset lineage and to define PCCPs for those models.
To explore detailed regulatory updates and AI/ML guidance, check out the Atlas Compliance blog.