
In regulated industries, non-conformance is a risk multiplier. One overlooked deviation, one undocumented procedure, or one inconsistent record can cascade into warning letters, failed inspections, or halted production lines. And the scrutiny is only intensifying; the FDA issued 561 Form 483s in FY 2024 alone, more than double what was issued just three years prior.
The message is clear: regulators are watching, and non-conformance is often where they look first. This guide is built for teams that can’t afford to operate on hindsight. We’ll break down what non-conformance really means, how to distinguish minor from major issues, and the steps your organization should take to detect, report, and prevent recurrence.
More importantly, you’ll learn how to shift from reactive compliance to predictive oversight before regulators do it for you.
Key Takeaways
- Non-conformance is your first red flag: Ignore it, and it turns into regulatory fire.
- Internal miss vs. regulatory breach: Non-conformance breaks your SOPs; non-compliance breaks the law.
- The 9-step NC process isn’t red tape: It’s your audit survival playbook.
- Stop firefighting: Real-time alerts, clean documentation, and trained teams keep you two steps ahead.
- Smart tech = smarter compliance: Platforms like Atlas turn chaos into control, before the FDA walks in.
What Is Non-Conformance?
Non-conformance refers to a failure to meet defined requirements, be it a documented SOP, an internal standard, or a regulatory mandate like 21 CFR. In regulated industries, it typically shows up as process deviations, incomplete records, or uncontrolled changes.
While not all non-conformances are critical, all require timely detection, documentation, and correction. They’re early indicators of systemic risk and key to audit outcomes.
Common Terms and Variations (Non-conformance vs. Non-compliance)
Teams often blur the lines between non-conformance and non-compliance, but in regulated environments, the distinction is critical. Here’s how the two terms differ and why accurate classification matters.
Non-conformance: A failure to follow internal standards; e.g., missing a documented step, using outdated forms, or skipping required checks
Non-compliance: A breach of external regulations, such as FDA, EMA, or ICH requirements.
Non-conformance can lead to non-compliance if unresolved. Regulators often trace audit findings back to patterns of unaddressed internal deviations.
Why It Matters in a Compliance-First Environment?
In regulated operations, a single undocumented deviation can compromise product integrity or inspection readiness.
Proactive non-conformance tracking gives quality and compliance teams visibility into weak points before auditors point them out. It’s how leading organizations move from firefighting to risk control.
In fact, a recent industry finding revealed that, on average, 10% to 18% of deviation and non-conformance investigations remained overdue between January 2022 and June 2023. This kind of delay is exactly what auditors catch and what sets off FDA observations.
Also Read: Common FDA Violations and Warning Letters Explained
What Are the Root Causes of Non-Conformance?

Non-conformance doesn’t happen in isolation; it’s almost always a symptom of something deeper. Identifying the source is key to preventing recurrence and ensuring audit readiness. Below are some causes that can lead to non-conformance.
1. Inadequate Documentation or SOPs
When SOPs are outdated, unclear, or inconsistently followed, deviations become inevitable. Teams can’t execute what isn’t documented well, or at all. In FDA-regulated environments, this often shows up as unsigned batch records, missing training logs, or conflicting work instructions.
2. Process Gaps or Inefficiencies
Processes that lack defined ownership, checkpoints, or built-in controls leave room for error. Whether it’s uncontrolled change management or poor batch handoffs, these inefficiencies often surface during audits, as repeat observations or CAPAs.
3. Weak Management Oversight or Commitment
If leadership treats compliance as a checkbox exercise, quality culture suffers. This shows up in poor deviation closure rates, delayed investigations, and reactive, not preventive, CAPA planning. Consistent oversight and visible support from management are essential for sustained conformance.
4. Communication Failures Across Teams
When quality, production, and regulatory affairs operate in silos, critical information gets missed. A failed deviation handoff or an unreported procedural change can quickly cascade into non-conformance. Cross-functional clarity is operational risk control.
What Are the Types of Non-Conformance?
Not all non-conformances carry the same risk. Classifying them correctly, especially during investigations or audits, is critical to determining response time, escalation, and documentation.
1. Minor Non-Conformance
A minor non-conformance is a low-risk deviation that doesn’t directly impact product quality, safety, or data integrity. These issues typically reflect isolated lapses or procedural misses that can be corrected without regulatory reporting.
Examples:
- A completed form missing one field (e.g., operator initials).
- Use of a superseded version of a log sheet with no impact on data.
- A calibration label is slightly misaligned but still readable and within the due date.
2. Major Non-Conformance
A major non-conformance indicates a failure that could affect product safety, efficacy, patient health, or regulatory obligations. These deviations often require immediate containment, full root cause analysis, and formal CAPA.
Examples:
- Untrained personnel performing critical tasks in GMP zones.
- Missing temperature data for controlled storage with no backup logs.
- Manufacturing step skipped and not documented until batch review.
How to Identify and Report Non-Conformance?
Early detection and structured reporting are the backbone of any compliant quality system. Whether triggered by audits, metrics, or frontline feedback, non-conformance must be flagged, documented, and routed for timely resolution.
1. Monitoring KPIs and Deviations
Tracking real-time metrics like deviation rates, CAPA closure times, and repeat observations helps flag emerging issues before they escalate. Quality teams often use dashboards tied to QMS or MES platforms to detect process drift and non-standard activity.
2. Internal/External Audits and Observations
Audits, whether scheduled or surprise, remain a primary channel for uncovering non-conformance. Internal audits expose process gaps early; external audits, including FDA or notified body inspections, often surface issues related to documentation, training, or control lapses. The FDA Establishment Inspection Report (EIR) reveals exactly what FDA inspectors flag and how missed non-conformances noted here can trigger future warning letters.
3. Customer Complaints and Feedback
Complaints often point to post-market failures or overlooked process risks. A single unresolved issue can lead to warning letters or product recalls, making formal tracking and escalation procedures non-negotiable. Mature systems link complaints to non-conformance reports and CAPAs.
How to Draft a Non-Conformance Report?
A strong non-conformance report ensures traceability, supports root cause analysis, and prepares your site for inspection readiness. Every report should include:
- Issue Description (date, time, location, equipment involved)
- Classification (minor/major, product/process/data)
- Immediate Actions Taken (containment or isolation steps)
- Supporting Evidence (logs, batch records, photos)
- Linked SOPs (what was expected vs. what occurred)
- Assigned Owner and Timeline for Investigation
In need of a more structured way to manage non-conformance reports? Platforms like Atlas-Compliance.ai simplify the process with built-in digital workflows, traceability, and audit-ready records, making documentation easier to manage, even in complex environments.
What Does the Non-Conformance Management Process Look Like?
Managing non-conformance is about driving a structured, repeatable process that addresses root causes, reduces recurrence, and stands up to regulatory scrutiny. Below is a consolidated view of each step in the NC management lifecycle.
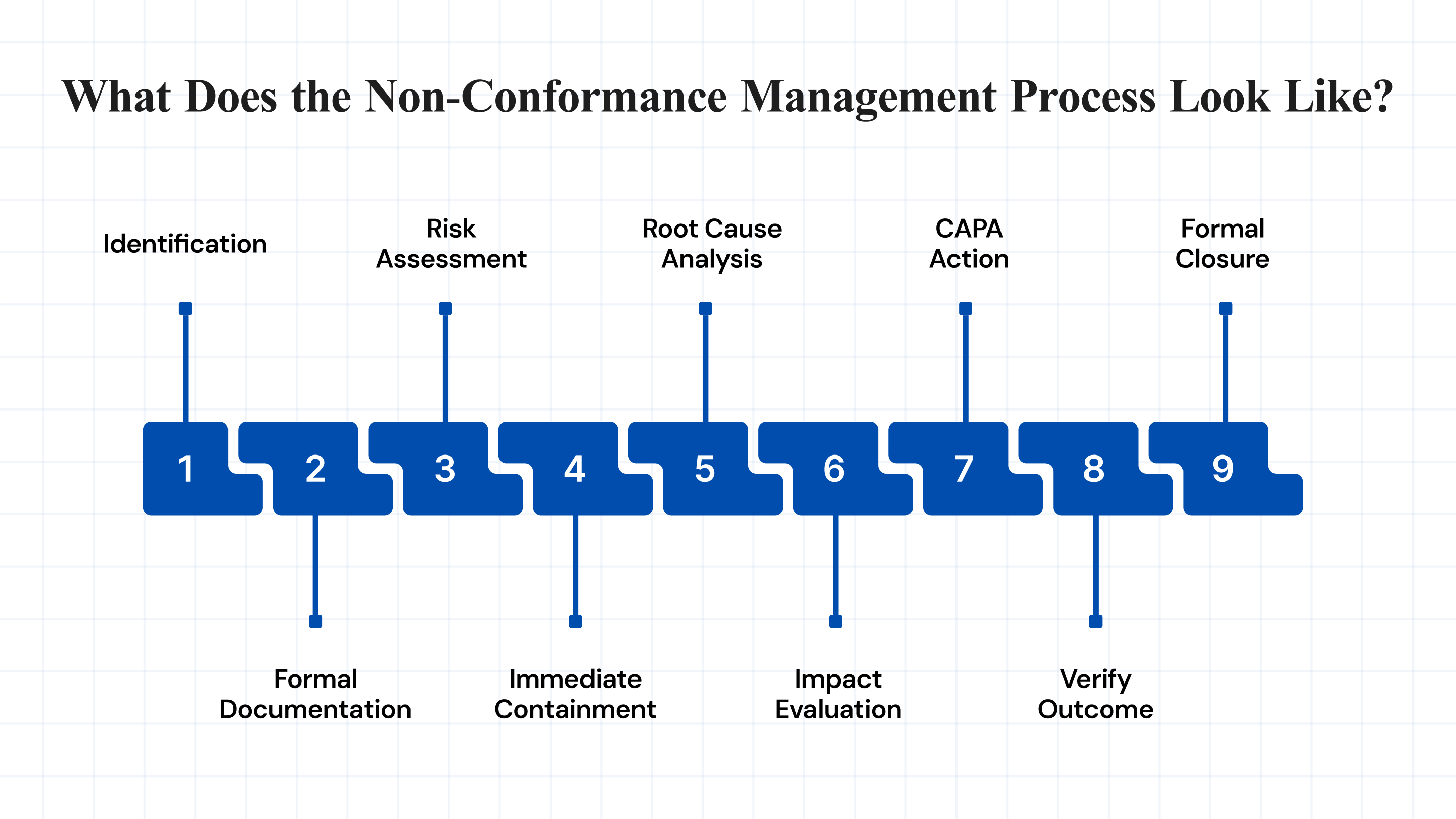
Snapshot of the Non-Conformance Management Process:
| Step | What Happens |
| 1. Identification | Issue detected via audit, alerts, or frontline reporting. |
| 2. Formal Documentation | Logged in QMS with who, what, where, and when. |
| 3. Risk Assessment | Classified as minor or major; urgency flagged. |
| 4. Immediate Containment | Immediate fix, hold product, isolate risk. |
| 5. Root Cause Analysis | Investigate why it happened, tools like 5 Whys or Ishikawa. |
| 6. Impact Check | Assess scope, affected batches, systems, or compliance exposure. |
| 7. CAPA Action | Fix root cause + prevent recurrence. |
| 8. Verify Outcome | Test if CAPA worked, data or audit check. |
| 9. Formal Closure | All actions done, documented, and filed. |
1. Identification
Non-conformance often starts with a signal, maybe a deviation, a failed batch, an audit observation, or even a customer complaint. Early detection allows teams to limit risk exposure and start documentation before the issue escalates. Mature QMS platforms like Atlas often integrate alerts and dashboards to catch these early.
2. Formal Documentation
Once flagged, the incident must be recorded in a centralized system. This includes the what, when, where, and by whom, ideally linked to batch records, equipment logs, or SOPs. Clear documentation ensures traceability and supports FDA inspection readiness.
3. Risk Assessment
Not every deviation carries the same weight. Is it a minor paperwork miss or a major GMP breach? Proper classification, based on impact to product quality, patient safety, or data integrity, guides urgency, escalation, and whether the FDA needs to be notified.
4. Immediate Containment
Containment is about damage control. The goal is to isolate affected materials, halt the process if needed, and prevent distribution. It’s not the fix, it’s the first response. Documentation here often includes quarantine logs, material holds, or temporary instructions to teams.
5. Root Cause Analysis
Surface-level fixes don’t hold. Using structured tools like 5 Whys, fault tree, or fishbone diagrams, teams trace back the issue to its origin. A rushed or shallow RCA risks recurrence and red flags during regulatory inspections.
6. Impact Evaluation
Now the team assesses how far the issue has spread. Did it affect just one batch, or was it systemic? Are there downstream quality implications? This step may involve trending historical data or rechecking previously approved records.
7. CAPA Action
Corrective and Preventive Actions aim to fix the issue and stop it from repeating. This may involve rewriting SOPs, retraining staff, adjusting equipment, or updating software. A strong CAPA plan is risk-ranked, time-bound, and assigned to accountable owners.
8. Verify Outcome
Before closing the loop, teams must confirm that the CAPA worked. This could mean retesting, sampling, or running a follow-up internal audit. FDA reviewers will look for evidence that the fix was implemented and effective.
9. Formal Closure
Only after verification is successful and documentation is complete can the NC be officially closed. Closure also involves linking the event to SOP updates, audit trails, and management review records, ensuring learnings are embedded into future operations.
How Can Compliance Teams Prevent Non-Conformance?
Preventing non-conformance requires embedding smarter controls into daily operations. Below are six practices that help quality and compliance teams reduce risk, improve response time, and stay ahead of FDA scrutiny:
- Run Internal Audits Frequently: Detect process gaps before inspectors do. Schedule risk-based audits tied to product criticality, past deviations, and compliance trends.
- Enable Real-Time Alerts & Monitoring: Use QMS tools that flag deviations the moment they occur. This allows rapid containment and supports proactive compliance.
- Train Teams on Updated SOPs: Outdated or ignored procedures are among the top FDA 483 observations. Ensure every role is trained, retrained, and documented.
- Evaluate Vendors and Suppliers Periodically: Non-conformance often originates outside your facility. Use structured vendor audits to check compliance readiness across your supply chain. If suppliers fail to meet requirements, teams often escalate using formal SCAR processes.
- Improve Documentation Hygiene: Standardize formats, ensure version control, and avoid gaps in signatures, timestamps, or approvals. Clean records are your first defense during inspections.
- Promote Continuous Improvement: Use CAPA trends and audit findings to refine processes. Embed improvement reviews into management meetings, not just after something goes wrong.
How Do Tools and Technology Support Non-Conformance Management?
Technology is becoming central to how regulated industries prevent, track, and resolve non-conformance. Here’s how key tools streamline your quality workflows:
1. QMS Software and Digital Workflows
Modern QMS platforms eliminate manual handoffs, standardize investigation workflows, and ensure audit trails are always complete. Integrated modules for deviations, CAPA, and training reduce delays and improve accountability.
2. Automated Alerts and Escalation Triggers
Real-time alerts help catch deviations as they happen. With rule-based triggers, teams can auto-flag major incidents, escalate unresolved CAPAs, or notify QA leads when high-risk vendors are involved.
3. Document Control Systems
From SOP updates to change control, digital DMS ensures version accuracy, traceability, and audit readiness. Look for systems that support electronic signatures, FDA 21 CFR Part 11 compliance, and linked training records.
Also Read: Understanding Electronic Document Management Systems (EDMS)
4. CAPA Tracking and Integration
Disconnected CAPAs lead to compliance gaps. An integrated system connects root cause analysis to CAPA action plans, tracks completion timelines, and links outcomes to future audits or training modules.
While various tools can support compliance workflows, Atlas Compliance brings them together into one intelligent platform, built specifically for regulated industries.
How Atlas Compliance Helps You Stay Audit-Ready?
Inspection prep shouldn’t begin when the audit letter arrives. With Atlas Compliance, you get the tools and insights to monitor risk in real time, long before issues escalate.
Here’s how the platform helps:
- AI-powered risk detection helps you spot compliance gaps early, not after they appear in a Form 483.
- Access the industry’s most comprehensive FDA intelligence database, including 483s, warning letters, and historical inspection patterns.
- Use real-time dashboards to track CAPA closure, SOP training, and documentation status, all in one place.
- Benchmark your compliance posture against industry peers to prioritize improvements that matter.
Whether you’re responding to an FDA observation or building a proactive quality system, Atlas helps you stay one step ahead, every time. Sign up today to get started!
Conclusion: Make Non-Conformance a Strategic Advantage
Non-conformance is a powerful signal. One that, if handled reactively, can spiral into repeat FDA observations, delayed submissions, and risk to safety. But when managed proactively, it becomes a powerful lens for improving processes, strengthening oversight, and building audit resilience.
In regulated industries, inspection readiness is non-negotiable. Whether you’re facing unannounced audits, global inspections, or mounting documentation requirements, your ability to detect, correct, and prevent non-conformance determines how inspection-ready you truly are.
Atlas Compliance gives you the visibility, automation, and insight needed to stay ahead, not just compliant. From automated alerts to FDA pattern analysis, we help quality and regulatory teams shift from manual reporting to smart, predictive control.
Book a demo today to streamline non-conformance and stay audit-ready, every day.
Frequently Asked Questions
What is an example of a nonconformance?
A nonconformance could be using an outdated version of an SOP during a manufacturing run or failing to calibrate equipment as per scheduled intervals. These lapses deviate from documented procedures or quality standards, potentially leading to regulatory risks, product quality issues, or FDA observations.
What should a company do if a nonconformity occurs?
The company should initiate containment measures immediately to prevent further impact, document the issue in a Non-Conformance Report (NCR), and perform a root cause analysis. Based on findings, corrective and preventive actions (CAPA) should be implemented and tracked until closure, with effectiveness checks to avoid recurrence.
What should a non-conformance report include?
A well-documented NCR should include a clear description of the issue, date of occurrence, affected process or product, potential impact, root cause findings, containment actions, and proposed CAPA. It should also list responsible personnel and link to relevant SOPs or inspection data to ensure traceability and accountability.
What two actions should immediately be taken when a non-conformance is noted?
First, contain the issue, isolate impacted products, processes, or documents to prevent further deviation. Second, log the non-conformance in the tracking system or NCR form to ensure proper documentation, visibility, and timely investigation. Early action helps limit compliance risk and supports faster CAPA turnaround.
How do you deal with non-conformance?
Managing non-conformance requires a structured process: identification, documentation, root cause analysis, containment, CAPA planning, and closure verification. Using tools like QMS software or platforms like Atlas Compliance streamlines this workflow, ensuring teams act quickly, maintain audit readiness, and prevent recurrence through proactive monitoring and alerts.